For Understanding and Controlling Complex System
Understand Protein Folding Before It Happens
Symbolic Protein Folding & Mutation Intelligence for predicting structural change, mutation impact, and folding behavior—without brute-force simulation.
For Understanding and Controlling Complex System
Symbolic Protein Folding & Mutation Intelligence for predicting structural change, mutation impact, and folding behavior—without brute-force simulation.
0%
Ubiquitin
Pass Rate
TM ≈ 0.9
0%
BRCA1 / BRCA2
Pass Rate
Stable Symbolic Seperation
0%
KRAS
Pass Rate
Mutation-sensitive validation
0%
Cross-Family
Pass Rate
High structural discrimination
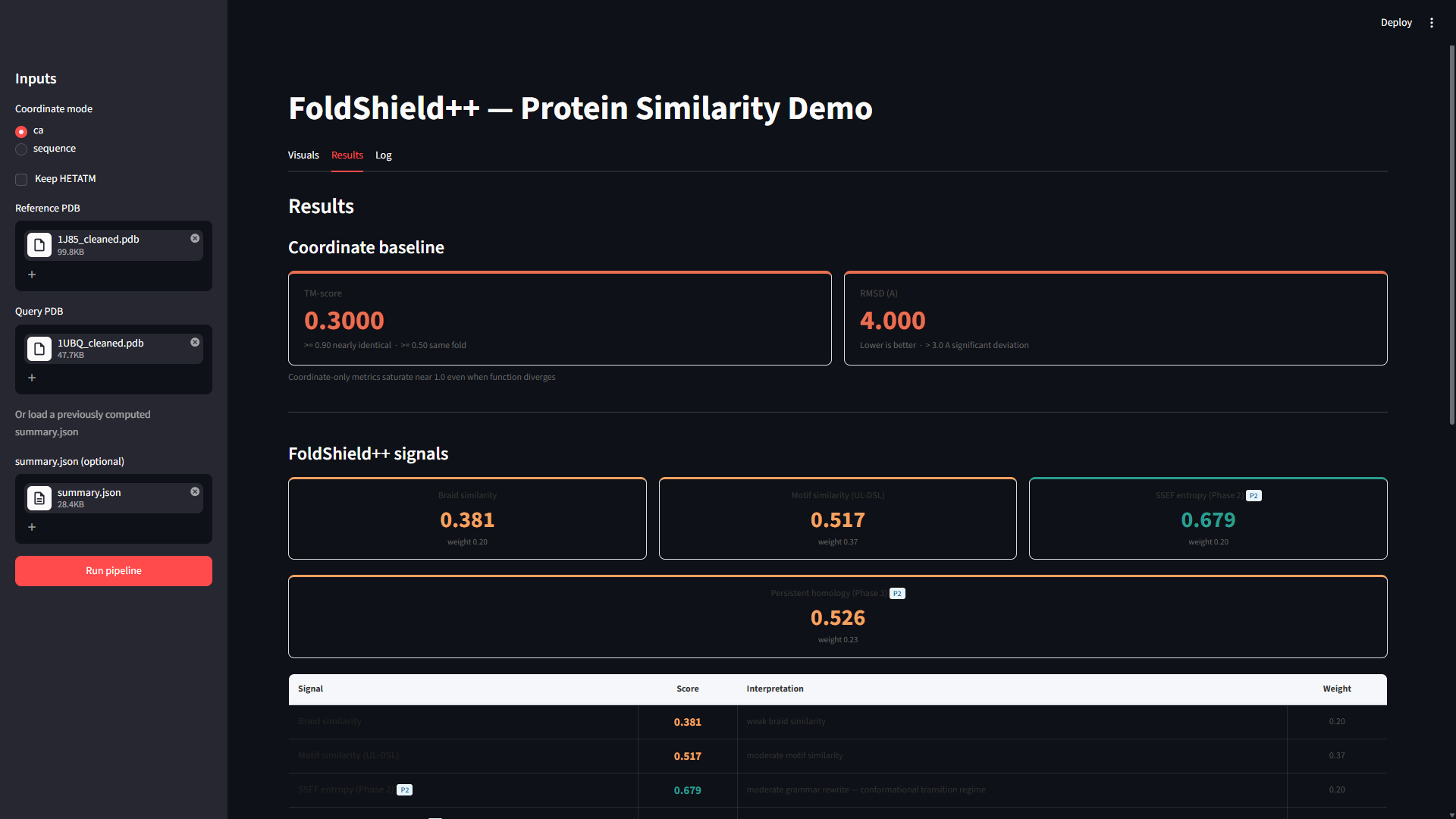
| Category | TM-Score | Behavioral | Symbolic Topology |
|---|---|---|---|
| Highly Similar | ≈ 0.9+ | 0.5 - 0.7 | 1.0 |
| Moderate | Moderate-High | Moderate | Stable |
| Dissimilar | 0.2 - 0.3 | 0.1 - 0.3 | ≈ 0.1 |
What This Validates
Structural similarity preserved
Mutation impact remains interpretable
Cross-family separation maintained
Symbolic topology remains stable
Folding behavior captured without MD simulation
CORE CAPABILITIES
The exponential growth in protein structure predictions—driven by AlphaFold2 and ESMFold—has created an urgent need for analysis tools that can scale beyond traditional geometric comparisons. Conventional metrics such as Root Mean Square Deviation (RMSD), Template Modeling Score (TM-score), and DALI alignments, while foundational, suffer from critical limitations: sensitivity to minor deviations, inability to capture dynamic behaviors, and poor interpretability in clinical and regulatory contexts.
Variant Interpretation & Functional Impact Prediction
01FoldShield++ identifies damaging variants by analyzing entropic and symbolic divergence — not just geometry. It detects subtle disruptions in structural dynamics that TM-score or RMSD cannot capture. This capability is vital for clinically relevant genes including BRCA1/2, KRAS, and TP53 where geometry alone fails to explain pathogenicity.

Homology, Analogy & Fold Classification
02Beyond geometric folds, FoldShield++ classifies proteins using topological invariants and symbolic motifs, enabling separation of homologs from analogs and supporting large-scale annotation of predicted proteomes.
Dynamics Interpretation Without MD Simulations
03Patchwise entropy correlation provides a cost-effective approximation of dynamic behavior, identifying conformational changes, flexible domains, activation states, and allosteric shifts without expensive molecular dynamics simulations.
Drug Discovery & Binder Design
04Entropy signatures reveal functional hotspots, while symbolic motifs identify conserved structural logic. These insights accelerate binder design, drug targeting, and variant-resistant therapeutic development.
Biotech Automation & Structural QC
05FoldShield++ provides automated quality-control layers for AlphaFold2/ESMFold outputs, detecting anomalous regions, fold instabilities, and low-confidence symbolic patterns at scale.
Symbolic AI Integration (UL-DSL, SMEA, QPhase)
06A distinguishing feature of FoldShield++ is transformation of biological structures into computational abstractions: proteins become symbolic programs via SynBraid, mutations become algebraic operations in SMEA, and structural reasoning becomes logical inference in UL-DSL.
Explainable AI for Structural Biology
07FoldShield++ delivers fine-grained, interpretable explanations for structural similarity, addressing queries such as: "Which structural regions diverged?" and "How did a mutation alter dynamics?
Massive-Scale Structure Comparison
08The discrete, compressible nature of symbolic and topological encodings enables efficient searching and clustering of millions of structures.
General Structural Intelligence System
09Fundamentally, FoldShield++ synthesizes geometric, topological, and entropic signals into a unified intelligence framework for structural biology.
FAQs
What is Foldshield++?
The exponential growth in protein structure predictions—driven by AlphaFold2 and ESMFold—has created an urgent need for analysis tools that can scale beyond traditional geometric comparisons. Conventional metrics such as Root Mean Square Deviation (RMSD), Template Modeling Score (TM-score), and DALI alignments, while foundational, suffer from critical limitations: sensitivity to minor deviations, inability to capture dynamic behaviors, and poor interpretability in clinical and regulatory contexts.